
Científicos del University College London, Reino Unido, estudian los enfoques de Machine Learning (ML) que han cerrado con éxito la brecha cada vez mayor entre grandes cantidades de secuencias de proteínas producidas por métodos de secuenciación de última generación y anotaciones de las estructuras y funciones de proteínas
(cbirt)-Uno de los mejores métodos para la predicción de estructuras, AlphaFold2 (AF2) , ha dado como resultado un aumento de 1000 veces en los datos estructurales de las proteínas, una hazaña notable en bioinformática estructural. Estos métodos ultrarrápidos pueden encontrar relaciones entre proteínas que no se podían encontrar con métodos anteriores. Esto significa que los científicos pueden descubrir más fácilmente cómo se relacionan las diferentes proteínas entre sí, incluso si no son muy similares.
Las proteínas juegan papeles esenciales en todos los procesos celulares. La predicción de la estructura de proteínas es fundamental para comprender las funciones de las proteínas. Los científicos se han preguntado acerca de cómo predecir la estructura de la proteína únicamente a partir de la secuencia de aminoácidos desde que se determinaron las primeras estructuras de proteínas globulares hace más de 50 años.
Actualmente, el Banco de datos de proteínas (PDB) informa 100 000 estructuras de proteínas únicas, que se determinaron experimentalmente utilizando técnicas experimentales de biología estructural como NMR, CryoEM y cristalografía de rayos X. Sin embargo, la gran cantidad de secuencias de proteínas generadas, en miles de millones, en los experimentos metagenómicos actuales superan en gran medida las estructuras conocidas.
Las proteínas están formadas por unidades estructurales llamadas dominios. Se sabe en gran medida que los dominios se pliegan de forma independiente y, por lo general, se asocian con roles funcionales específicos. Por lo tanto, comprender la estructura del dominio de la proteína y el plegamiento son cruciales para determinar las funciones de la proteína.
Numerosas bases de datos como SCoP, CATH y ECOD han documentado y clasificado estructuras de dominio y doblado grupos en superfamilias evolutivas. Estas bases de datos han clasificado hasta 5000 estructuras de dominio y alrededor de 1300 pliegues. Los estudios han revelado que estos números están saturados, y son las diversas combinaciones de dominios las que dan como resultado una gran cantidad de funciones de proteínas. Por lo tanto, comprender las combinaciones de dominios y la evolución es de suma importancia para descubrir las funciones de varias proteínas no caracterizadas.
Con el advenimiento de los enfoques de Machine Learning en biología estructural, AlphaFold (AF) ha mejorado drásticamente la precisión de la predicción de la estructura de la proteína utilizando redes neuronales profundas. Junto con el aprendizaje profundo (DL), el método también utiliza restricciones evolutivas, físicas y geométricas en las estructuras de proteínas.
AlphaFold se desempeñó extremadamente bien en la Evaluación crítica de la predicción de estructuras (CASP13). Varios grupos de investigación intentaron replicar el método debido a que al principio no estaba disponible para la comunidad científica. RoseTTAFold y PREFMD lanzaron nuevas versiones que competían con AlphaFold, pero AlphaFold2 en CASP14 superó a todos los demás métodos. Se dice que la base de datos de estructuras de proteínas AlphaFold (AFDB) tiene 214 millones de estructuras de proteínas putativas disponibles, que abarcan todo UniProt, según lo anunciado por DeepMind. También se dice que estas supuestas estructuras proteicas están disponibles en la plataforma 3D-Beacons del Instituto Europeo de Bioinformática (EBI).
Enfoques basados en secuencias para encontrar homólogos
Los métodos de inferencia basada en homología (HBI) se han utilizado con éxito para transferir anotaciones de proteínas marcadas a proteínas sin marcar de secuencia similar. Aparte de estos, los alineamientos de secuencias múltiples (MSA) son un almacén de información evolutiva, que a menudo se utilizan en técnicas para determinar estructuras de proteínas de novo y funciones de proteínas. Sin embargo, estos métodos se ven obstaculizados por tiempos de ejecución lentos y parámetros defectuosos, lo que genera MSA poco informativos. Si bien la computación avanzada ha resuelto en gran medida los problemas de tiempo de ejecución, las bases de datos de secuencias cada vez mayores requieren métodos mejores y más avanzados.
Los pLM y los embebidos como alternativa
Los modelos de lenguaje del Procesamiento del Lenguaje Natural (NLP) demuestran ser una alternativa efectiva a los métodos HBI. Incorporar técnicas de aprendizaje profundo (DL) para aprender la ‘gramática’ del ‘lenguaje’ de la vida codificado en miles de millones de secuencias de proteínas conocidas utilizando la configuración del modelo de lenguaje de NLP. Los pLM (modelos de lenguaje de proteínas) aprenden implícitamente atributos de datos como restricciones evolutivas, funcionales y estructurales en secuencias de proteínas, a diferencia de los modelos ML típicos. Se aplican la autorregresión y el aprendizaje de lenguaje enmascarado para implementar el aprendizaje autosupervisado de pLM. La autorregresión implica un entrenamiento basado en la predicción de un resultado futuro (en este caso, token) basado en todos los resultados anteriores (tokens anteriores). El aprendizaje de lenguaje enmascarado implica la reconstrucción de secuencias corruptas (tokens) a partir de tokens no corruptos.
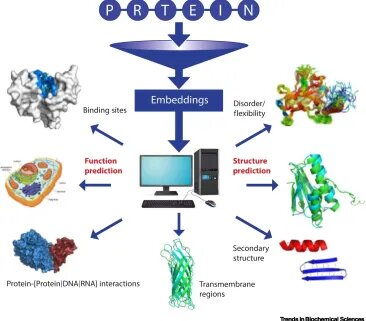
La repetición del entrenamiento con miles de millones de secuencias de proteínas da como resultado que el pLM aprenda las propiedades estadísticas del ‘lenguaje’. Esto se puede lograr extrayendo incrustaciones, los estados ocultos de las redes neuronales (pLM). Si bien el entrenamiento de modelos es computacionalmente intensivo y generalmente requiere instalaciones informáticas de alto rendimiento, la extracción de incrustaciones se puede lograr incluso en PC o portátiles.
Se ha demostrado que los pLM mejoran con respecto a HBI en la predicción de las funciones de las proteínas. La transferencia de anotación basada en incrustación (EAT) compara las proteínas en el espacio de incrustación en lugar del espacio de secuencia para los métodos HBI. En Evaluación crítica de anotación funcional (CAFA4), EAT superó significativamente a HBI.
Se ha demostrado que la anotación basada en estructuras (SAT) captura proteínas evolutivamente distantes de manera más eficiente que EAT o HBI. Los avances recientes en la forma de AlphaFold2 en la predicción de estructuras de proteínas de calidad de rayos X han llevado a nuevas aplicaciones para los métodos SAT en biología estructural. Los estudios que involucran el análisis de estructuras de proteínas de la AFDB utilizando algoritmos de reconocimiento de pliegues han dado como resultado el descubrimiento de nuevos pliegues de proteínas.
Se sabe que los alineadores estructurales tradicionales son ineficientes para mantenerse al día con la gran cantidad de bases de datos cada vez mayores de estructuras de proteínas. Están surgiendo nuevos alineadores estructurales utilizando el enfoque basado en secuencias, como Foldseek. Las incrustaciones de pLM, junto con alineadores estructurales rápidos, podrían abarcar vastas regiones del espacio de pliegue de proteínas en términos de clasificación y validación de asignaciones.
Los autores analizaron los dominios estructurales predichos en AlphaFold2 para 21 organismos modelo. El análisis condujo a la identificación de 2367 nuevas familias.
La disponibilidad de AlphaFold2 como herramienta junto con AFDB acelerará notablemente los proyectos de investigación que anteriormente se retrasaron debido a la falta de estructuras de proteínas de calidad derivadas experimentalmente. Hay advertencias que hay que tener en cuenta. No se abordan las mutaciones puntuales y las variantes de un solo aminoácido. La sobrerrepresentación de los estados de plegamiento de proteínas da como resultado un modelo no representativo para otros estados alternativos.
Conclusión
Recientemente, las técnicas de aprendizaje profundo de vanguardia han transformado el panorama de la investigación de proteínas. El método AlphaFold2 basado en DL aumenta drásticamente el repertorio estructural de proteínas de alta calidad con una precisión muy alta. El uso de modelos de lenguaje previamente entrenados permite una mejor anotación y predicción que los métodos HBI. Las herramientas de alineación estructural junto con los pLM pueden atravesar mejor el espacio de proteínas para predecir relaciones evolutivas distantes. En otras palabras, cuando se encuentran con enfoques de ML, los métodos de bioinformática estructural conducen a métodos mejores, más rápidos y más avanzados para la determinación de la estructura y la función de las proteínas. Estos nuevos enfoques de ML tienen el potencial de mejorar significativamente nuestra comprensión de las proteínas y su papel en la biología y la medicina.

