El software DeepMind que puede predecir la forma 3D de las proteínas ya está cambiando la biología
(nature)- Durante más de una década, el biólogo molecular Martin Beck y sus colegas han intentado armar uno de los rompecabezas más difíciles del mundo: un modelo detallado de la máquina molecular más grande en las células humanas.
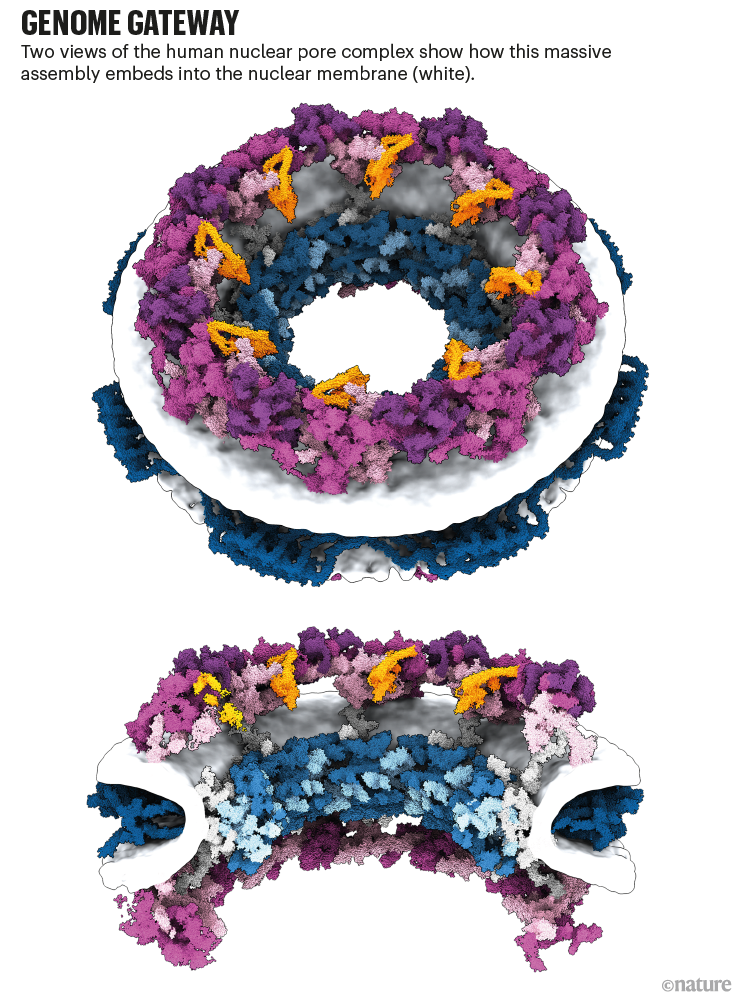
Este gigante, llamado complejo de poros nucleares, controla el flujo de moléculas dentro y fuera del núcleo de la célula, donde se encuentra el genoma. Cientos de estos complejos existen en cada célula. Cada uno está compuesto por más de 1000 proteínas que juntas forman anillos alrededor de un agujero a través de la membrana nuclear.
Estas 1000 piezas de rompecabezas se extraen de más de 30 bloques de construcción de proteínas que se entrelazan de innumerables maneras. Para hacer el rompecabezas aún más difícil, las formas 3D determinadas experimentalmente de estos bloques de construcción son un popurrí de estructuras reunidas de muchas especies, por lo que no siempre encajan bien. Y la imagen en la caja del rompecabezas, una vista en 3D de baja resolución del complejo de poros nucleares, carece de detalles suficientes para saber cuántas de las piezas encajan con precisión.
En 2016, un equipo dirigido por Beck, con sede en el Instituto Max Planck de Biofísica (MPIB) en Frankfurt, Alemania, informó un modelo 1 que cubría aproximadamente el 30% del complejo de poros nucleares y alrededor de la mitad de los 30 bloques de construcción. llamadas proteínas Nup.
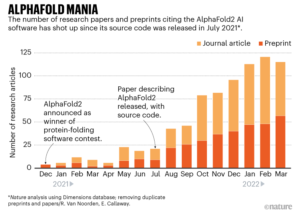
Luego, en julio pasado, la firma londinense DeepMind, parte de Alphabet, la empresa matriz de Google, hizo pública una herramienta de inteligencia artificial (IA) llamada AlphaFold 2 . El software podría predecir la forma 3D de las proteínas a partir de su secuencia genética, en su mayor parte, con una precisión milimétrica. Esto transformó la tarea de Beck y los estudios de miles de otros biólogos (ver ‘manía AlphaFold’).
“AlphaFold cambia el juego”, dice Beck. “Esto es como un terremoto. Puedes verlo en todas partes”, dice Ora Schueler-Furman, bióloga estructural computacional de la Universidad Hebrea de Jerusalén en Israel, que está utilizando AlphaFold para modelar interacciones de proteínas. “Hay un antes de julio y un después”.
Usando AlphaFold, Beck y otros en el MPIB, la bióloga molecular Agnieszka Obarska-Kosinska y un grupo dirigido por el bioquímico Gerhard Hummer, así como un equipo dirigido por el modelador estructural Jan Kosinski, en el Laboratorio Europeo de Biología Molecular (EMBL) en Hamburgo, Alemania. , podría predecir las formas de las versiones humanas de las proteínas Nup con mayor precisión. Y aprovechando un ajuste que ayudó a AlphaFold a modelar cómo interactúan las proteínas, lograron publicar un modelo en octubre pasado que cubría el 60 % del complejo 3 . Revela cómo el complejo estabiliza los agujeros en el núcleo, además de insinuar cómo el complejo controla lo que entra y sale.
En el último medio año, la manía AlphaFold se ha apoderado de las ciencias de la vida. “En cada reunión en la que participo, la gente dice ‘¿por qué no usar AlphaFold?’”, dice Christine Orengo, bióloga computacional del University College London.
En algunos casos, la IA ha ahorrado tiempo a los científicos; en otros, ha hecho posible investigaciones que antes eran inconcebibles o muy poco prácticas. Tiene limitaciones, y algunos científicos encuentran que sus predicciones son demasiado poco confiables para su trabajo. Pero el ritmo de experimentación es frenético.
Incluso aquellos que desarrollaron el software luchan por mantenerse al día con su uso en áreas que van desde el descubrimiento de fármacos y el diseño de proteínas hasta los orígenes de la vida compleja. “Me despierto y escribo AlphaFold en Twitter”, dice John Jumper, quien dirige el equipo de AlphaFold en DeepMind. “Es toda una experiencia ver todo.”
Un éxito sorprendente
AlphaFold causó sensación en diciembre de 2020 , cuando dominó un concurso llamado Evaluación crítica de la predicción de la estructura de proteínas, o CASP. La competencia, que se lleva a cabo cada dos años, mide el progreso en uno de los mayores desafíos de la biología: determinar las formas 3D de las proteínas solo a partir de su secuencia de aminoácidos. Las entradas de software de computadora se juzgan contra las estructuras de las mismas proteínas determinadas usando métodos experimentales como la cristalografía de rayos X o la microscopía crioelectrónica (crio-EM), que disparan rayos X o haces de electrones a las proteínas para construir una imagen de su forma.
La versión 2020 de AlphaFold fue la segunda edición del software. También ganó el CASP de 2018 , pero sus esfuerzos anteriores en su mayoría no fueron lo suficientemente buenos como para reemplazar estructuras determinadas experimentalmente, dice Jumper. Sin embargo, las predicciones de AlphaFold2 estaban, en promedio, a la par con las estructuras empíricas.
No estaba claro cuándo DeepMind haría que el software o sus predicciones estuvieran ampliamente disponibles, por lo que los investigadores utilizaron la información de una charla pública de Jumper y sus propios conocimientos para desarrollar su propia herramienta de inteligencia artificial, llamada RoseTTAFold .
Luego, el 15 de julio de 2021, aparecieron artículos que describían RoseTTAFold y AlphaFold2 2 , 4 , junto con código de código abierto disponible gratuitamente y otra información necesaria para que los especialistas ejecuten sus propias versiones de las herramientas. Una semana después, DeepMind anunció que había utilizado AlphaFold para predecir la estructura de casi todas las proteínas fabricadas por humanos , así como los ‘proteomas’ completos de otros 20 organismos ampliamente estudiados, como ratones y la bacteria Escherichia coli : más de 365 000 estructuras en total (ver ‘Lo que se sabe sobre los proteomas’). DeepMind también los publicó públicamente en una base de datos mantenida por el Instituto Europeo de Bioinformática del EMBL.(EMBL–EBI), en Hinxton, Reino Unido. Desde entonces, esa base de datos se ha incrementado a casi un millón de estructuras.
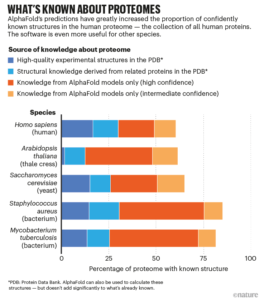
Este año, DeepMind planea lanzar un total de más de 100 millones de predicciones de estructuras. Eso es casi la mitad de todas las proteínas conocidas, y cientos de veces más que la cantidad de proteínas determinadas experimentalmente en el depósito de estructuras del Protein Data Bank (PDB).
AlphaFold implementa redes neuronales de aprendizaje profundo: arquitecturas computacionales inspiradas en el cableado neuronal del cerebro para discernir patrones en los datos. Ha sido entrenado en cientos de miles de estructuras y secuencias de proteínas determinadas experimentalmente en el PDB y otras bases de datos. Ante una nueva secuencia, primero busca secuencias relacionadas en las bases de datos, que pueden identificar aminoácidos que han tendido a evolucionar juntos, lo que sugiere que están cerca en el espacio 3D. La estructura de las proteínas relacionadas existentes proporciona otra forma de estimar las distancias entre los pares de aminoácidos en la nueva secuencia.
AlphaFold itera las pistas de estas pistas paralelas de un lado a otro mientras intenta modelar las posiciones 3D de los aminoácidos, actualizando continuamente su estimación. Los especialistas dicen que la aplicación del software de nuevas ideas en la investigación del aprendizaje automático parece ser lo que hace que AlphaFold sea tan bueno; en particular, el uso de un mecanismo de inteligencia artificial denominado «atención» para determinar qué conexiones de aminoácidos son las más destacadas para su tarea en cualquier momento.
La confianza de la red en la información sobre secuencias de proteínas relacionadas significa que AlphaFold tiene algunas limitaciones. No está diseñado para predecir el efecto de mutaciones, como las que causan enfermedades, en la forma de una proteína. Tampoco fue entrenado para determinar cómo las proteínas cambian de forma en presencia de otras proteínas que interactúan o moléculas como las drogas. Pero sus modelos vienen con puntajes que miden la confianza de la red en su predicción para cada unidad de aminoácido de una proteína, y los investigadores están modificando el código de AlphaFold para expandir sus capacidades.
Hasta ahora, más de 400.000 personas han utilizado la base de datos AlphaFold de EMBL-EBI, según DeepMind. También hay ‘usuarios avanzados’ de AlphaFold: investigadores que configuraron el software en sus propios servidores o recurrieron a versiones de AlphaFold basadas en la nube para predecir estructuras que no están en la base de datos EMBL-EBI, o idear nuevos usos para la herramienta. .
Resolver estructuras
Los biólogos ya están impresionados con la capacidad de AlphaFold para resolver estructuras. “Según lo que he visto hasta ahora, confío bastante en AlphaFold”, dice Thomas Boesen, biólogo estructural de la Universidad de Aarhus en Dinamarca. El software ha predicho con éxito las formas de las proteínas que el centro de Boesen ha determinado pero que aún no ha publicado. “Esa es una gran validación de mi parte”, dice. Él y la ecologista microbiana de Aarhus, Tina Šantl-Temkiv, están utilizando AlphaFold para modelar la estructura de las proteínas bacterianas que promueven la formación de hielo, y que podrían contribuir a los efectos de enfriamiento del hielo en las nubes, porque los biólogos no han podido determinar completamente el estructuras experimentalmente 5 .
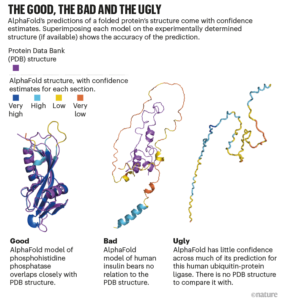
Siempre que una proteína se enrolle en una sola forma 3D bien definida, y no todas lo hacen, la predicción de AlphaFold puede ser difícil de superar, dice Arne Elofsson, bioinformático de proteínas de la Universidad de Estocolmo. «Es una solución de un solo clic para obtener probablemente el mejor modelo que obtendrá».
Donde AlphaFold tiene menos confianza, «es muy bueno para decirle cuando no funciona», dice Elofsson. En tales casos, las estructuras predichas pueden parecerse a hilos de espagueti flotantes (ver ‘Lo bueno, lo malo y lo feo’). Esto a menudo corresponde a regiones de proteínas que carecen de una forma definida, al menos de forma aislada. Tales regiones intrínsecamente desordenadas, que constituyen alrededor de un tercio del proteoma humano, podrían definirse bien solo cuando otra molécula, como un compañero de señalización, está presente.
Norman Davey, biólogo computacional del Instituto de Investigación del Cáncer de Londres, dice que la capacidad de AlphaFold para identificar trastornos ha cambiado las reglas del juego para su trabajo de estudio de las propiedades de estas regiones. “Al instante hubo un gran aumento en la calidad de las predicciones que teníamos, sin ningún esfuerzo de nuestra parte”, dice.
El volcado de estructuras de proteínas de AlphaFold en la base de datos EMBL-EBI también se está poniendo en uso de inmediato. El equipo de Orengo lo está buscando para identificar nuevos tipos de proteínas (sin verificarlas experimentalmente) y ha encontrado cientos, quizás miles, de familias de proteínas potencialmente nuevas, ampliando el conocimiento de los científicos sobre cómo se ven y qué pueden hacer las proteínas. En otro esfuerzo, el equipo está revisando bases de datos de secuencias de ADN recolectadas del océano y aguas residuales, para tratar de identificar nuevas enzimas que se alimentan de plástico. Usando AlphaFold para aproximar rápidamente las estructuras de miles de proteínas, los investigadores esperan comprender mejor cómo evolucionaron las enzimas para descomponer el plástico y, potencialmente, mejorarlas.
Sergey Ovchinnikov, biólogo evolutivo de la Universidad de Harvard en Cambridge, Massachusetts, dice que la capacidad de transformar cualquier secuencia de genes codificadores de proteínas en una estructura confiable debería ser especialmente poderosa para los estudios de evolución. Los investigadores comparan secuencias genéticas para determinar cómo se relacionan los organismos y sus genes entre especies. Para los genes relacionados de forma lejana, las comparaciones podrían fallar en encontrar parientes evolutivos debido a que las secuencias han cambiado mucho. Pero al comparar las estructuras de proteínas, que tienden a cambiar menos rápidamente que las secuencias genéticas, los investigadores podrían descubrir relaciones antiguas pasadas por alto. “Esto abre una oportunidad increíble para estudiar la evolución de las proteínas y los orígenes de la vida”, dice Pedro Beltrao, biólogo computacional del Instituto Federal Suizo de Tecnología en Zúrich.
Para probar esta idea, un equipo dirigido por Martin Steinegger, biólogo computacional de la Universidad Nacional de Seúl, y sus colegas utilizaron una herramienta que desarrollaron, llamada Foldseek, para buscar parientes de la enzima copiadora de ARN del SARS-CoV-2, la virus que causa el COVID-19 — en la base de datos AlphaFold de EMBL-EBI 6 . Esta búsqueda arrojó posibles parientes antiguos no identificados previamente: proteínas en eucariotas, incluidos los mohos mucilaginosos, que se asemejan, en su estructura 3D, a enzimas llamadas transcriptasas inversas que los virus como el VIH usan para copiar el ARN en ADN, a pesar de que tienen muy poca similitud en el aspecto genético. nivel de secuencia.
Asistente experimental
Para los científicos que desean determinar la estructura detallada de una proteína específica, una predicción AlphaFold no es necesariamente una solución inmediata. Más bien, proporciona una aproximación inicial que puede validarse o refinarse mediante experimentos, y que en sí mismo ayuda a dar sentido a los datos experimentales. Los datos sin procesar de la cristalografía de rayos X, por ejemplo, aparecen como patrones de rayos X difractados. Por lo general, los científicos necesitan una conjetura inicial sobre la estructura de una proteína para interpretar estos patrones. Anteriormente, a menudo improvisaban información de proteínas relacionadas en el PDB o usaban enfoques experimentales, dice Randy Read, biólogo estructural de la Universidad de Cambridge, Reino Unido, cuyo laboratorio se especializó en algunos de estos métodos. Ahora, las predicciones de AlphaFold han hecho que tales enfoques sean innecesarios para la mayoría de los patrones de rayos X, dice Read, y su laboratorio está trabajando para hacer un mejor uso de AlphaFold en modelos experimentales. “Hemos reenfocado totalmente nuestra investigación”.
Él y otros investigadores han utilizado AlphaFold para determinar estructuras cristalinas a partir de datos de rayos X que no se podían interpretar sin un modelo de partida adecuado. “La gente está resolviendo estructuras que, durante años, no habían sido resueltas”, dice Claudia Millán Nebot, ex postdoctorado en el laboratorio de Read que ahora trabaja en la empresa de análisis SciBite en Cambridge. Ella espera ver un exceso de nuevas estructuras de proteínas enviadas al PDB, en gran parte como resultado de AlphaFold.
Lo mismo ocurre con los laboratorios que se especializan en crio-EM, que captura imágenes de proteínas ultracongeladas. En algunos casos, los modelos de AlphaFold han predicho con precisión características únicas de proteínas llamadas receptores acoplados a proteína G (GPCR, por sus siglas en inglés), que son objetivos importantes de fármacos, que otras herramientas computacionales fallaron, dice Bryan Roth, biólogo estructural y farmacólogo de la Universidad de Carolina del Norte en Chapel Hill. “Parece ser realmente bueno para generar los primeros modelos, que luego refinamos con algunos datos experimentales”, dice. “Eso nos ahorra algo de tiempo”.
Pero Roth agrega que AlphaFold no siempre es tan preciso. De las varias docenas de estructuras GPCR que su laboratorio ha resuelto, pero que aún no se han publicado, dice, «aproximadamente la mitad de las veces, las estructuras AlphaFold son bastante buenas y la mitad de las veces son más o menos inútiles para nuestros propósitos». En algunos casos, dice, AlphaFold etiqueta las predicciones con un alto nivel de confianza, pero las estructuras experimentales muestran que es incorrecto. Incluso cuando el software lo hace bien, no puede modelar cómo se vería una proteína cuando se une a un fármaco u otra molécula pequeña (ligando), lo que puede alterar sustancialmente la estructura. Tales advertencias hacen que Roth se pregunte cuán útil será AlphaFold para el descubrimiento de fármacos .
Es cada vez más común en los esfuerzos de descubrimiento de fármacos utilizar software de acoplamiento computacional que analiza miles de millones de moléculas pequeñas para encontrar algunas que puedan unirse a las proteínas, una indicación de que podrían fabricar fármacos útiles. Roth ahora está trabajando con Brian Shoichet, un químico médico de la Universidad de California, San Francisco, para ver cómo las predicciones de AlphaFold se comparan con las estructuras determinadas experimentalmente en este ejercicio.
Shoichet dice que están limitando su trabajo a las proteínas para las cuales la predicción de AlphaFold concuerda con las estructuras experimentales. Pero incluso en estos casos, el software de acoplamiento muestra diferentes aciertos de drogas para la estructura experimental y la toma de AlphaFold, lo que sugiere que las pequeñas discrepancias podrían ser importantes. “Eso no significa que no encontraremos nuevos ligandos, simplemente encontraremos otros diferentes”, dice Shoichet. Su equipo ahora está sintetizando fármacos potenciales identificados mediante estructuras AlphaFold y probando su actividad en el laboratorio.
Optimismo crítico
Los investigadores de las empresas farmacéuticas y biotecnológicas están entusiasmados con el potencial de AlphaFold para ayudar en el descubrimiento de fármacos, dice Shoichet. “Optimismo crítico es como lo describiría”. En noviembre de 2021, DeepMind lanzó su propio spin-off, IsoMorphic Labs , cuyo objetivo es aplicar AlphaFold y otras herramientas de IA al descubrimiento de fármacos. Pero la compañía ha dicho poco más sobre sus planes.
Karen Akinsanya, quien lidera el desarrollo terapéutico en Schrödinger, una firma de descubrimiento de fármacos con sede en la ciudad de Nueva York que también publica software de simulación química, dice que ella y sus colegas ya están teniendo cierto éxito al usar estructuras AlphaFold, incluso para GPCR, en pantallas virtuales y diseño de compuestos para candidatos a fármacos. Ella encuentra que, al igual que con las estructuras experimentales, se necesita software adicional para llegar a los detalles finos de las cadenas laterales de aminoácidos o las ubicaciones donde podrían sentarse los átomos de hidrógeno individuales. Una vez hecho esto, las estructuras AlphaFold han demostrado ser lo suficientemente buenas como para guiar el descubrimiento de fármacos, en algunos casos.
“Es difícil decir ‘esto es una panacea’; que debido a que puede hacerlo muy bien para una estructura, sorprendente y emocionantemente bien, es eminentemente aplicable a todas las estructuras. Claramente no lo es”, dice Akinsanya. Y ella y sus colegas descubrieron que las predicciones de precisión de AlphaFold no muestran si una estructura será útil para la detección de drogas posterior. Las estructuras AlphaFold nunca reemplazarán por completo a las experimentales en el descubrimiento de fármacos, dice. Pero podrían acelerar el proceso al complementar los métodos experimentales.
Los desarrolladores de fármacos curiosos por AlphaFold recibieron buenas noticias en enero, cuando DeepMind levantó una restricción clave sobre su uso para aplicaciones comerciales . Cuando la compañía lanzó el código de AlphaFold en julio de 2021, había estipulado que los parámetros, o pesos, necesarios para ejecutar la red neuronal AlphaFold, el resultado final de entrenar la red en cientos de miles de estructuras y secuencias de proteínas, eran para fines no comerciales. Usar unicamente. Akinsanya dice que esto fue un cuello de botella para algunos en la industria, y hubo una «ola de emoción» cuando DeepMind cambió de rumbo. (RoseTTAFold vino con restricciones similares, dice Ovchinnikov, uno de sus desarrolladores. Pero la próxima versión será completamente de código abierto).
Las herramientas de IA no solo están cambiando la forma en que los científicos determinan cómo se ven las proteínas. Algunos investigadores los están utilizando para hacer proteínas completamente nuevas. “El aprendizaje profundo está transformando por completo la forma en que se realiza el diseño de proteínas en mi grupo”, dice David Baker, bioquímico de la Universidad de Washington en Seattle y líder en el campo del diseño de proteínas, así como en la predicción de sus estructuras. Su equipo, con el químico computacional Minkyung Baek, dirigió el trabajo para desarrollar RoseTTAFold.
El equipo de Baker obtiene AlphaFold y RoseTTAFold para «alucinar» nuevas proteínas. Los investigadores han alterado el código de IA para que, dadas secuencias aleatorias de aminoácidos, el software los optimice hasta que se parezcan a algo que las redes neuronales reconozcan como una proteína (ver ‘Soñando con proteínas’).
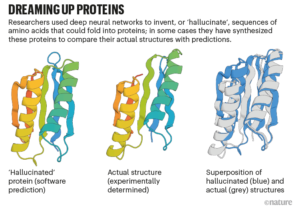
En diciembre de 2021, Baker y sus colegas informaron que expresaron 129 de estas proteínas alucinadas en bacterias y descubrieron que aproximadamente una quinta parte de ellas se plegaban en algo parecido a su forma predicha 7 . “Esa es realmente la primera demostración de que se pueden diseñar proteínas usando estas redes”, dice Baker. Su equipo ahora está utilizando este enfoque para diseñar proteínas que hacen cosas útiles, como catalizar una reacción química particular, especificando los aminoácidos responsables de la función deseada y dejando que la IA invente el resto.
Hackear AlphaFold
Cuando DeepMind lanzó su código AlphaFold, Ovchinnikov quería comprender mejor cómo funcionaba la herramienta. En cuestión de días, él y sus colegas de biología computacional, incluido Steinegger, crearon un sitio web llamado ColabFold que permitía a cualquier persona enviar una secuencia de proteínas a AlphaFold o RoseTTAFold y obtener una predicción de la estructura. Ovchinnikov imaginó que él y otros científicos utilizarían ColabFold para intentar «romper» AlphaFold, por ejemplo, proporcionando información falsa sobre los parientes evolutivos de una secuencia de proteína objetivo. Al hacer esto, Ovchinnikov esperaba poder determinar cómo la red había aprendido a predecir estructuras tan bien.
Al final resultó que, la mayoría de los investigadores que usaron ColabFold solo querían obtener una estructura de proteína. Pero otros lo usaron como plataforma para modificar las entradas de AlphaFold para abordar nuevas aplicaciones. “No esperaba la cantidad de ataques de varios tipos”, dice Jumper.
Con mucho, el truco más popular ha sido manejar la herramienta en complejos de proteínas compuestos por múltiples cadenas de péptidos que interactúan y, a menudo, se entrelazan. Al igual que con el complejo del poro nuclear, muchas proteínas en las células adquieren su función cuando forman complejos con múltiples subunidades de proteínas.
AlphaFold fue diseñado para predecir la forma de cadenas peptídicas individuales, y su entrenamiento consistió completamente en dichas proteínas. Pero la red parece haber aprendido algo sobre cómo se juntan los complejos. Varios días después de que se publicara el código de AlphaFold, Yoshitaka Moriwaki, un bioinformático de proteínas de la Universidad de Tokio, tuiteó que podía predecir con precisión las interacciones entre dos secuencias de proteínas si se unían con una secuencia enlazadora larga. Baek pronto compartió otro truco para predecir complejos, obtenido del desarrollo de RoseTTAFold.
ColabFold luego incorporó la capacidad de predecir complejos. Y en octubre de 2021, DeepMind lanzó una actualización llamada AlphaFold-Multimer 8 que se entrenó específicamente en complejos de proteínas, a diferencia de su predecesor. El equipo de Jumper lo aplicó a miles de complejos en el PDB y descubrió que predecía alrededor del 70 % de las interacciones proteína-proteína conocida.
Estas herramientas ya están ayudando a los investigadores a detectar posibles nuevos socios proteicos. El equipo de Elofsson utilizó AlphaFold para predecir las estructuras de 65.000 pares de proteínas humanas que se sospechaba que interactuaban sobre la base de datos experimentales 9 . Y un equipo dirigido por Baker usó AlphaFold y RoseTTAFold para modelar las interacciones entre casi todos los pares de proteínas codificadas por la levadura, identificando más de 100 complejos previamente desconocidos 10 . Tales pantallas son solo puntos de partida, dice Elofsson. Hacen un buen trabajo al predecir algunos emparejamientos de proteínas, particularmente aquellos que son estables, pero tienen dificultades para identificar interacciones más transitorias. “Que se vea bien no significa que sea correcto”, dice Elofsson. “Necesitas algunos datos experimentales que demuestren que tienes razón”.
El trabajo del complejo de poros nucleares es un buen ejemplo de cómo las predicciones y los datos experimentales pueden trabajar juntos, dice Kosinski (ver ‘Puerta de enlace del genoma’). «No es que tomemos las 30 proteínas, las arrojemos a AlphaFold y saquemos la estructura». Para juntar las estructuras proteicas predichas, el equipo usó imágenes 3D del complejo del poro nuclear, capturadas usando una forma de crio-EM llamada tomografía crioelectrónica . En un caso, los experimentos que pueden determinar la proximidad de las proteínas revelaron una interacción sorprendente entre dos componentes del complejo, que luego confirmaron los modelos de AlphaFold.
Kosinski ve el mapa actual del complejo de poros nucleares del equipo como un punto de partida para experimentos y simulaciones que examinan cómo funciona el complejo de poros y cómo funciona mal en la enfermedad.
Los límites de AlphaFold
A pesar de todo el progreso realizado con AlphaFold, los científicos dicen que es importante tener claras sus limitaciones, particularmente porque los investigadores que no se especializan en predecir estructuras de proteínas lo usan.
Los intentos de aplicar AlphaFold a varias mutaciones que interrumpen la estructura natural de una proteína, incluida una relacionada con el cáncer de mama temprano, han confirmado que el software no está equipado para predecir las consecuencias de nuevas mutaciones en las proteínas, ya que no hay secuencias relacionadas con la evolución para examinar. 11 _
El equipo de AlphaFold ahora está pensando en cómo se podría diseñar una red neuronal para hacer frente a nuevas mutaciones. Jumper espera que esto requiera que la red prediga mejor cómo una proteína pasa de su estado desplegado a su estado plegado. Eso probablemente necesitaría un software que se base solo en lo que ha aprendido sobre la física de proteínas para predecir estructuras, dice Mohammed AlQuraishi, biólogo computacional de la Universidad de Columbia en la ciudad de Nueva York. “Algo que nos interesa es hacer predicciones a partir de secuencias individuales sin usar información evolutiva”, dice. “Ese es un problema clave que permanece abierto”.
AlphaFold también está diseñado para predecir una sola estructura, aunque ha sido pirateado para escupir más de una. Pero muchas proteínas adquieren múltiples conformaciones, lo que puede ser importante para su función. “AlphaFold realmente no puede lidiar con proteínas que pueden adoptar diferentes estructuras en diferentes conformaciones”, dice Schueler-Furman. Y las predicciones son para estructuras aisladas, mientras que muchas proteínas funcionan junto con ligandos como el ADN y el ARN, moléculas de grasa y minerales como el hierro. «Todavía nos faltan ligandos, nos falta todo lo demás sobre las proteínas», dice Elofsson.
Desarrollar estas redes neuronales de próxima generación será un gran desafío, dice AlQuraishi. AlphaFold se basó en décadas de investigación que generaron estructuras experimentales de proteínas de las que la red podría aprender. Ese volumen de datos actualmente no está disponible para capturar la dinámica de las proteínas o las formas de los trillones de moléculas más pequeñas con las que las proteínas podrían interactuar. El PDB incluye estructuras de proteínas a medida que interactúan con otras moléculas, pero esto captura solo una pequeña parte de la diversidad química, agrega Jumper.
Los investigadores creen que les llevará tiempo determinar la mejor manera de manejar AlphaFold y las herramientas de IA relacionadas. AlQuraishi ve paralelismos con los primeros días de la televisión, cuando algunos programas consistían en emisoras de radio que simplemente leían las noticias. «Creo que vamos a encontrar nuevas aplicaciones de estructura que aún no hemos concebido».
Dónde terminará la revolución AlphaFold es una incógnita. “Las cosas están cambiando tan rápido”, dice Baker. “Incluso en el próximo año, vamos a ver avances realmente importantes con el uso de estas herramientas”. Janet Thornton, bióloga computacional del EMBL-EBI, cree que uno de los mayores impactos de AlphaFold podría ser simplemente convencer a los biólogos para que estén más abiertos a los conocimientos de los enfoques teóricos y computacionales. “Para mí, la revolución es el cambio de mentalidad”, dice.
La revolución AlphaFold ha inspirado a Kosinski a soñar en grande. Él imagina que las herramientas inspiradas en AlphaFold podrían usarse para modelar no solo proteínas y complejos individuales, sino también orgánulos completos o incluso células hasta el nivel de moléculas de proteína individuales. “Este es el sueño que seguiremos en las próximas décadas”.

