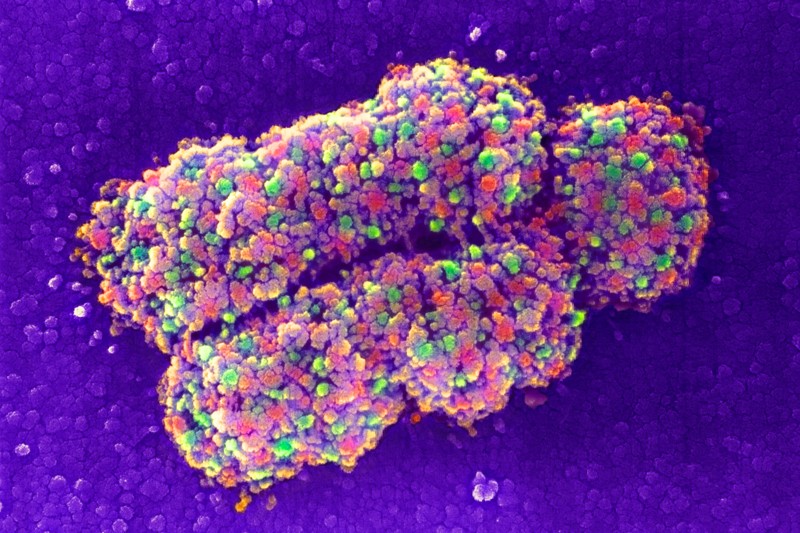
Desde la edición de genes hasta la determinación de la estructura de proteínas y la computación cuántica, aquí hay siete tecnologías que probablemente tendrán un impacto en la ciencia
Por Miguel Eisenstein
(Nature)- Nuestro quinto resumen anual de las herramientas que parecen destinadas a revolucionar la ciencia este año:
Genomas completamente terminados
Aproximadamente una décima parte del genoma humano permaneció sin cartografiar cuando los investigadores de genómica Karen Miga de la Universidad de California, Santa Cruz, y Adam Phillippy del Instituto Nacional de Investigación del Genoma Humano en Bethesda, Maryland, lanzaron el consorcio Telomere-to-Telomere (T2T). en 2019. Ahora, ese número se ha reducido a cero. En una preimpresión publicada en mayo del año pasado, el consorcio informó sobre la primera secuencia de extremo a extremo del genoma humano, agregando casi 200 millones de nuevos pares de bases a la secuencia del genoma de consenso humano ampliamente utilizada conocida como GRCh38, y escribiendo el capítulo final de el Proyecto Genoma Humano 1 .
Lanzado por primera vez en 2013, GRCh38 ha sido una herramienta valiosa: un andamio en el que mapear lecturas de secuenciación. Pero está plagado de agujeros. Esto se debe en gran parte a que la tecnología de secuenciación ampliamente utilizada desarrollada por Illumina, en San Diego, California, produce lecturas precisas, pero cortas. No son lo suficientemente largos para mapear sin ambigüedades secuencias genómicas altamente repetitivas, incluidos los telómeros que cubren los extremos de los cromosomas y los centrómeros que coordinan la partición del ADN recién replicado durante la división celular.
Las tecnologías de secuenciación de lectura larga demostraron cambiar las reglas del juego. Desarrolladas por Pacific Biosciences en Menlo Park, California, y Oxford Nanopore Technologies (ONT) en Oxford, Reino Unido, estas tecnologías pueden secuenciar decenas o incluso cientos de miles de bases en una sola lectura, pero, al menos al principio, no sin errores. . Para cuando el equipo de T2T reconstruyó 2 , 3sus primeros cromosomas individuales, X y 8, en 2020, sin embargo, la secuenciación de Pacific Biosciences había avanzado hasta el punto de que los científicos de T2T podían detectar pequeñas variaciones en largos tramos de secuencias repetidas. Estas «huellas dactilares» sutiles hicieron que los segmentos cromosómicos largos y repetitivos fueran manejables, y el resto del genoma rápidamente se alineó. La plataforma ONT también captura muchas modificaciones en el ADN que modulan la expresión génica, y T2T también pudo mapear estas «etiquetas epigenéticas» en todo el genoma 4 .
El genoma T2T resuelto procedía de una línea celular que contiene dos conjuntos idénticos de cromosomas. Los genomas humanos diploides normales contienen dos versiones de cada cromosoma, y los investigadores ahora están trabajando en estrategias de «fases» que pueden asignar con confianza cada secuencia a la copia cromosómica apropiada. “Ya estamos obteniendo algunos ensamblajes por fases bastante fenomenales”, dice Miga.
Este trabajo de ensamblaje diploide se lleva a cabo en colaboración con la organización socia de T2T, el Consorcio de Referencia del Pangenoma Humano, que aspira a producir un mapa del genoma más representativo, basado en cientos de donantes de todo el mundo. “Nuestro objetivo es capturar un promedio del 97 % de la diversidad alélica humana”, dice Erich Jarvis, uno de los principales investigadores del consorcio y genetista de la Universidad Rockefeller en la ciudad de Nueva York. Como presidente del Vertebrate Genomes Project, Jarvis también espera aprovechar estas capacidades de ensamblaje completo del genoma para generar secuencias completas para cada especie de vertebrado en la Tierra. “Creo que dentro de los próximos 10 años, estaremos haciendo genomas de telómero a telómero de manera rutinaria”, dice.
Soluciones de estructuras proteicas
La estructura dicta la función. Pero puede ser difícil de medir. Los importantes avances experimentales y computacionales de los últimos dos años han brindado a los investigadores herramientas complementarias para determinar estructuras de proteínas con una velocidad y resolución sin precedentes.
El algoritmo de predicción de estructuras AlphaFold2, desarrollado por DeepMind, filial de Alphabet en Londres, se basa en estrategias de «aprendizaje profundo» para extrapolar la forma de una proteína plegada a partir de su secuencia de aminoácidos 5 . Después de una victoria decisiva en la competencia Evaluación crítica de la predicción de la estructura de proteínas de 2020, en la que los biólogos computacionales prueban sus algoritmos de predicción de estructuras cara a cara, la reputación y la adopción de AlphaFold2 se han disparado. «Para algunas de las estructuras, las predicciones son casi inquietantemente buenas», dice Janet Thornton, científica principal y ex directora del Instituto Europeo de Bioinformática en Hinxton, Reino Unido. Desde su lanzamiento al público en julio pasado, AlphaFold2 se ha aplicado a proteomas para determinar las estructuras de todas las proteínas expresadas en humanos 6y en 20 organismos modelo (ver Nature 595 , 635; 2021 ), así como cerca de 440 000 proteínas en la base de datos Swiss-Prot, lo que aumenta considerablemente la cantidad de proteínas para las que hay datos de modelado de alta confianza disponibles. El algoritmo AlphaFold también ha demostrado su capacidad para abordar complejos de proteínas de cadena múltiple 7 .
Paralelamente, las mejoras en la microscopía electrónica criogénica (crio-EM) están permitiendo a los investigadores resolver experimentalmente incluso las proteínas y los complejos más desafiantes. Cryo-EM escanea moléculas congeladas con un haz de electrones, generando imágenes de las proteínas en múltiples orientaciones que luego se pueden volver a ensamblar computacionalmente en una estructura 3D. En 2020, las mejoras en el hardware y software crio-EM permitieron que dos equipos generaran estructuras con una resolución de menos de 1,5 ångströms, capturando la posición de átomos individuales 8 , 9. «Antes de esto, discutíamos el término ‘resolución atómica’ con desenfreno, pero solo ha sido casi atómico», dice Bridget Carragher, codirectora del Centro de Microscopía Electrónica Simons del Centro de Biología Estructural de Nueva York en la ciudad de Nueva York. “Esto realmente es atómico”. Y, aunque ambos equipos utilizaron una proteína modelo especialmente bien estudiada llamada apoferritina, dice Carragher, estos estudios sugieren que la resolución casi atómica también es factible para otros objetivos más difíciles.
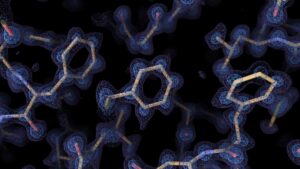
Muchos experimentadores que inicialmente se mostraron escépticos acerca de AlphaFold2 ahora lo ven como un claro complemento de los métodos experimentales como cryo-EM, donde sus modelos computacionales pueden ayudar en el análisis y la reconstrucción de datos. Y cryo-EM puede generar hallazgos que actualmente están fuera del alcance de la predicción computacional. El equipo de Carragher, por ejemplo, está utilizando crio-EM ‘resuelto en el tiempo’ para capturar cambios conformacionales rápidos que ocurren cuando las proteínas interactúan con otras moléculas. “Podemos atrapar cosas y ver lo que sucede en el orden de cien milisegundos”, dice ella.
También existe un entusiasmo considerable en torno a un método relacionado, la tomografía crioelectrónica (crio-ET), que captura el comportamiento natural de las proteínas en secciones delgadas de células congeladas. Pero la interpretación de estas imágenes abarrotadas y complicadas es un desafío, y Carragher cree que los avances computacionales del mundo del aprendizaje automático serán esenciales. “¿De qué otra manera vamos a resolver estos problemas casi intratables?” ella pregunta.
Simulación cuántica
Los átomos son, bueno, de tamaño atómico. Pero bajo las condiciones adecuadas, pueden ser inducidos a un estado de gran tamaño altamente excitado con diámetros del orden de un micrómetro o más. Al realizar esta excitación en conjuntos cuidadosamente colocados de cientos de átomos de manera controlada, los físicos han demostrado que pueden resolver problemas físicos desafiantes que llevan a las computadoras convencionales a sus límites.
Las computadoras cuánticas administran datos en forma de qubits. Combinados mediante el fenómeno de la física cuántica llamado entrelazamiento, los qubits pueden influirse entre sí a distancia. Estos qubits pueden aumentar drásticamente la potencia informática que se puede lograr con una asignación determinada de qubits en relación con una cantidad equivalente de bits en una computadora clásica.
Varios grupos han utilizado con éxito iones individuales como qubits, pero sus cargas eléctricas dificultan su ensamblaje a alta densidad. Físicos como Antoine Browaeys de la agencia de investigación nacional francesa CNRS en París y Mikhail Lukin de la Universidad de Harvard en Cambridge, Massachusetts, están explorando un enfoque alternativo. Los equipos utilizan pinzas ópticas para posicionar con precisión átomos sin carga en matrices 2D y 3D estrechamente empaquetadas, luego aplican láseres para excitar estas partículas en «átomos Rydberg» de gran diámetro que se enredan con sus vecinos 10 , 11. “Los sistemas atómicos de Rydberg se pueden controlar individualmente y sus interacciones se pueden activar y desactivar”, explica el físico Jaewook Ahn del Instituto Avanzado de Ciencia y Tecnología de Corea en Daejeon, Corea del Sur. Esto a su vez confiere programabilidad.
Este enfoque ha cobrado un impulso considerable en el lapso de unos pocos años, con avances tecnológicos que han mejorado la estabilidad y el rendimiento de las matrices atómicas de Rydberg, así como un escalado rápido de unas pocas docenas de qubits a varios cientos. Las primeras aplicaciones se han centrado en problemas definidos, como la predicción de las propiedades de los materiales, pero el enfoque es versátil. “Hasta ahora, cualquier modelo teórico que propusieron los teóricos, había una manera de implementarlo”, dice Browaeys.
Los pioneros en el campo han fundado empresas que están desarrollando sistemas basados en arreglos atómicos de Rydberg para uso en laboratorio, y Browaeys estima que tales simuladores cuánticos podrían estar disponibles comercialmente en uno o dos años. Pero este trabajo también podría allanar el camino hacia computadoras cuánticas que se pueden aplicar de manera más general, incluso en economía, logística y encriptación. Los investigadores aún luchan por definir el lugar de esta tecnología aún incipiente en el mundo de la computación, pero Ahn establece paralelismos con el impulso inicial de los hermanos Wright en la aviación. “Ese primer avión no tenía ninguna ventaja de transporte”, dice Ahn, “pero finalmente cambió el mundo”.
Manipulación precisa del genoma
A pesar de toda su destreza en la edición del genoma, la tecnología CRISPR-Cas9 es más adecuada para la inactivación de genes que para la reparación. Esto se debe a que, aunque dirigir la enzima Cas9 a una secuencia genómica es relativamente preciso, la reparación celular del corte de doble cadena resultante no lo es. Mediadas por un proceso llamado unión de extremos no homólogos, las reparaciones de CRISPR-Cas9 a menudo se ven enturbiadas por pequeñas inserciones o eliminaciones.
La mayoría de las enfermedades genéticas requieren la corrección de genes en lugar de la interrupción, señala David Liu, biólogo químico de la Universidad de Harvard en Cambridge. Liu y su equipo han desarrollado dos enfoques prometedores para hacer precisamente eso. Ambos explotan la orientación precisa de CRISPR al mismo tiempo que limitan la capacidad de Cas9 para cortar el ADN en ese sitio. La primera, llamada edición de bases, acopla una forma catalíticamente alterada de Cas9 a una enzima que ayuda a la conversión química de un nucleótido a otro, por ejemplo, citosina a timina o adenina a guanina (ver Nature https://doi.org/hc2t; 2016). Pero actualmente solo se puede acceder a ciertos cambios de base a base con este método. La edición principal, el desarrollo más reciente del equipo, vincula a Cas9 con un tipo de enzima conocida como transcriptasa inversa y utiliza un ARN guía que se modifica para incluir la edición deseada de la secuencia genómica (ver Nature 574 , 464–465; 2019 ). A través de un proceso bioquímico de varias etapas, estos componentes copian el ARN guía en ADN que finalmente reemplaza la secuencia del genoma objetivo. Es importante destacar que tanto la edición básica como la principal cortan solo una hebra de ADN, un proceso más seguro y menos disruptivo para las células.
Descrita por primera vez en 2016, la edición básica ya está en camino a la clínica: Beam Therapeutics, fundada por Liu y también con sede en Cambridge, recibió el visto bueno en noviembre de la Administración de Drogas y Alimentos de EE. UU. para probar este enfoque en humanos por primera vez. con el objetivo de reparar el gen que causa la enfermedad de células falciformes.
La edición principal no está tan avanzada, pero continúan surgiendo iteraciones mejoradas, y la promesa del método es clara. Hyongbum Henry Kim, especialista en edición del genoma de la Facultad de Medicina de la Universidad de Yonsei en Seúl, y su equipo han demostrado que pueden lograr hasta un 16 % de eficiencia utilizando la edición principal para corregir mutaciones genéticas en la retina en ratones 12 . “Si usáramos versiones más avanzadas reportadas recientemente, las eficiencias mejorarían aún más”, dice. Y el grupo de Liu descubrió que la maquinaria principal puede ayudar a la inserción de secuencias de ADN del tamaño de un gen en el genoma, lo que podría ofrecer una estrategia más segura y más estrictamente controlada para la terapia génica 13. El proceso es relativamente ineficiente, pero incluso una pequeña reparación a veces puede ser muy útil, señala Liu. “En algunos casos, se sabe que si se puede reemplazar un gen en un nivel del 10 % o incluso del 1 %, se puede rescatar la enfermedad”, dice.
Terapias genéticas dirigidas
Los medicamentos basados en ácidos nucleicos pueden estar teniendo un impacto en la clínica, pero aún están muy limitados en cuanto a los tejidos en los que se pueden aplicar. La mayoría de las terapias requieren la administración local o la manipulación ex vivo de las células que se recolectan y luego se trasplantan nuevamente a un paciente. Una excepción destacada es el hígado, que filtra el torrente sanguíneo y está demostrando ser un objetivo sólido para la administración selectiva de fármacos. En este caso, la administración intravenosa, o incluso subcutánea, puede hacer el trabajo.
«Simplemente conseguir la entrega a cualquier tejido es difícil, cuando realmente piensas en el desafío», dice Daniel Anderson, ingeniero químico del Instituto de Tecnología de Massachusetts (MIT) en Cambridge. “Nuestros cuerpos están diseñados para usar la información genética que tenemos, no para aceptar a los recién llegados”. Pero los investigadores están logrando un progreso constante en el desarrollo de estrategias que pueden ayudar a guiar estos medicamentos a sistemas de órganos específicos mientras evitan otros tejidos que no son el objetivo.
Los virus adenoasociados son el vehículo de elección para muchos esfuerzos de terapia génica, y los estudios en animales han demostrado que la selección cuidadosa del virus correcto combinado con promotores de genes específicos de tejido puede lograr una entrega eficiente restringida a órganos 14 . Sin embargo, los virus a veces son difíciles de fabricar a escala y pueden provocar respuestas inmunitarias que socavan la eficacia o producen eventos adversos.
Las nanopartículas de lípidos brindan una alternativa no viral, y varios estudios publicados en los últimos años destacan el potencial para ajustar su especificidad. Por ejemplo, el enfoque de detección selectiva de órganos (SORT, por sus siglas en inglés) desarrollado por el bioquímico Daniel Siegwart y sus colegas en el Centro Médico Southwestern de la Universidad de Texas en Dallas, permite la generación y detección rápidas de nanopartículas lipídicas para identificar aquellas que pueden atacar de manera efectiva las células en tejidos como como el pulmón o el bazo 15. «Ese fue uno de los primeros artículos que mostró que si se hace una detección sistemática de estas nanopartículas de lípidos y se empieza a cambiar su composición, se puede sesgar la biodistribución», dice Roy van der Meel, ingeniero biomédico de la Universidad Tecnológica de Eindhoven en el Países Bajos. Varios grupos también están explorando cómo los componentes proteicos, como los anticuerpos específicos de células, podrían ayudar en el proceso de focalización, señala Anderson.
Anderson está particularmente entusiasmado con el progreso preclínico en la detección de precursores de células sanguíneas e inmunitarias en la médula ósea demostrado por compañías como Beam Therapeutics e Intellia en Cambridge, las cuales utilizan formulaciones especialmente diseñadas de nanopartículas de lípidos. El éxito en apuntar a esos tejidos, dice, podría ahorrarles a los pacientes el agotador proceso involucrado con las actuales terapias génicas ex vivo , que incluyen quimioterapia para destruir la médula ósea existente antes del trasplante. “Hacer estas cosas in vivo realmente podría cambiar el tratamiento de los pacientes”, dice Anderson.
Multiómica espacial
La explosión en el desarrollo de ómicas unicelulares significa que los investigadores ahora pueden derivar rutinariamente conocimientos genéticos, transcriptómicos, epigenéticos y proteómicos de células individuales, a veces simultáneamente (ver go.nature.com/3nnhooo ). Pero las técnicas unicelulares también sacrifican información crucial al arrancar estas células de sus entornos nativos.
En 2016, investigadores dirigidos por Joakim Lundeberg en el KTH Royal Institute of Technology de Estocolmo diseñaron una estrategia para superar este problema. El equipo preparó portaobjetos con oligonucleótidos con código de barras (hebras cortas de ARN o ADN) que pueden capturar el ARN mensajero de una rebanada de tejido intacto, de modo que cada transcrito podría asignarse a una posición particular en la muestra de acuerdo con su código de barras. «Nadie creía realmente que pudiéramos sacar un análisis de todo el transcriptoma de una sección de tejido», dice Lundeberg. “Pero resultó ser sorprendentemente fácil”.
Desde entonces, el campo de la transcriptómica espacial se ha disparado. Ahora hay varios sistemas comerciales disponibles, incluida la plataforma Visium Spatial Gene Expression de 10x Genomics, que se basa en la tecnología de Lundeberg. Los grupos académicos continúan desarrollando métodos innovadores que pueden mapear la expresión génica con una profundidad y resolución espacial cada vez mayores.
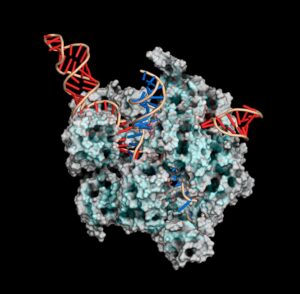
Ahora los investigadores están superponiendo más información ómica sobre sus mapas espaciales. Por ejemplo, el ingeniero biomédico Rong Fan de la Universidad de Yale en New Haven, Connecticut, desarrolló una plataforma conocida como DBiT-seq 16, que emplea un sistema de microfluidos que puede generar simultáneamente códigos de barras para miles de transcritos de ARNm y cientos de proteínas marcadas con anticuerpos marcados con oligonucleótidos. Esto puede proporcionar una evaluación mucho más precisa de cómo la expresión génica celular influye en la producción y la actividad de las proteínas que la que se podría obtener solo a partir de datos transcriptómicos, y el equipo de Fan lo ha estado utilizando para investigar procesos como la activación de células inmunitarias. “Estamos viendo los primeros signos de cómo las células inmunitarias de la piel reaccionan a la vacuna Moderna COVID-19”, dice. Algunos sistemas comerciales también pueden capturar datos espaciales de múltiples proteínas en paralelo con conocimientos transcriptómicos, incluida la plataforma Visium y el sistema GeoMx de Nanostring.
Mientras tanto, el grupo de Lundeberg ha refinado su método de transcriptómica espacial para capturar simultáneamente datos de secuencias de ADN. Esto ha permitido a su equipo comenzar a mapear los eventos espaciotemporales que subyacen a la tumorigénesis. “Podríamos seguir estos cambios genéticos en el espacio, cómo evolucionan hacia variantes genéticas adicionales que eventualmente conducen al tumor”, dice.
El equipo de Fan ha demostrado el mapeo espacial de las modificaciones de la cromatina en muestras de tejido, lo que puede revelar los paisajes reguladores de genes celulares que influyen en procesos como el desarrollo, la diferenciación y la comunicación intercelular 17 . Fan confía en que el método se puede combinar con el análisis espacial del ARN e incluso de las proteínas. “Tenemos datos preliminares que muestran que esto es totalmente factible”, dice.
Diagnóstico basado en CRISPR
La capacidad del sistema CRISPR-Cas para la escisión precisa de secuencias específicas de ácidos nucleicos se deriva de su papel como «sistema inmunitario» bacteriano contra la infección viral. Este enlace inspiró a los primeros usuarios de la tecnología a contemplar la aplicabilidad del sistema al diagnóstico viral. «Simplemente tiene mucho sentido usar para lo que están diseñados en la naturaleza», dice Pardis Sabeti, genetista del Instituto Broad del MIT y Harvard en Cambridge. “Tienes miles de millones de años de evolución de tu lado”.
Pero no todas las enzimas Cas son iguales. Cas9 es la enzima de referencia para la manipulación del genoma basada en CRISPR, pero gran parte del trabajo en el diagnóstico basado en CRISPR ha empleado la familia de moléculas dirigidas al ARN conocidas como Cas13, identificadas por primera vez en 2016 por el biólogo molecular Feng Zhang y su equipo en El ancho. «Cas13 utiliza su guía de ARN para reconocer un objetivo de ARN mediante el emparejamiento de bases y activa una actividad de ribonucleasa que puede aprovecharse como herramienta de diagnóstico mediante el uso de un ARN informador», explica Jennifer Doudna de la Universidad de California, Berkeley, quien compartió el Premio Nobel de Química 2020 con Emmanuelle Charpentier, ahora en la Unidad Max Planck para la Ciencia de los Patógenos en Berlín, por desarrollar las capacidades de edición del genoma de CRISPR-Cas9. Esto se debe a que Cas13 no solo corta el ARN objetivo del ARN guía, también realiza una ‘escisión colateral’ en cualquier otra molécula de ARN cercana. Muchos diagnósticos basados en Cas13 utilizan un ARN informador que une una etiqueta fluorescente a una molécula extintora que inhibe esa fluorescencia. Cuando Cas13 se activa después de reconocer el ARN viral, corta el indicador y libera la etiqueta fluorescente del extintor, generando una señal detectable. Algunos virus dejan una firma lo suficientemente fuerte como para lograr la detección sin amplificación, lo que simplifica el diagnóstico en el punto de atención. Por ejemplo, en enero pasado, Doudna y Melanie Ott del Gladstone Institute of Virology en San Francisco, California, demostraron una prueba rápida CRISPR-Cas13 basada en un hisopo nasal para la detección sin amplificación del SARS-CoV-2 usando un teléfono móvil. cámara Muchos diagnósticos basados en Cas13 utilizan un ARN informador que une una etiqueta fluorescente a una molécula extintora que inhibe esa fluorescencia. Cuando Cas13 se activa después de reconocer el ARN viral, corta el indicador y libera la etiqueta fluorescente del extintor, generando una señal detectable. Algunos virus dejan una firma lo suficientemente fuerte como para lograr la detección sin amplificación, lo que simplifica el diagnóstico en el punto de atención. Por ejemplo, en enero pasado, Doudna y Melanie Ott del Gladstone Institute of Virology en San Francisco, California, demostraron una prueba rápida CRISPR-Cas13 basada en un hisopo nasal para la detección sin amplificación del SARS-CoV-2 usando un teléfono móvil. cámara Muchos diagnósticos basados en Cas13 utilizan un ARN informador que une una etiqueta fluorescente a una molécula extintora que inhibe esa fluorescencia. Cuando Cas13 se activa después de reconocer el ARN viral, corta el indicador y libera la etiqueta fluorescente del extintor, generando una señal detectable. Algunos virus dejan una firma lo suficientemente fuerte como para lograr la detección sin amplificación, lo que simplifica el diagnóstico en el punto de atención. Por ejemplo, en enero pasado, Doudna y Melanie Ott del Gladstone Institute of Virology en San Francisco, California, demostraron una prueba rápida CRISPR-Cas13 basada en un hisopo nasal para la detección sin amplificación del SARS-CoV-2 usando un teléfono móvil. cámara corta el reportero y libera la etiqueta fluorescente del extintor, generando una señal detectable. Algunos virus dejan una firma lo suficientemente fuerte como para lograr la detección sin amplificación, lo que simplifica el diagnóstico en el punto de atención. Por ejemplo, en enero pasado, Doudna y Melanie Ott del Gladstone Institute of Virology en San Francisco, California, demostraron una prueba rápida CRISPR-Cas13 basada en un hisopo nasal para la detección sin amplificación del SARS-CoV-2 usando un teléfono móvil. cámara corta el reportero y libera la etiqueta fluorescente del extintor, generando una señal detectable. Algunos virus dejan una firma lo suficientemente fuerte como para lograr la detección sin amplificación, lo que simplifica el diagnóstico en el punto de atención. Por ejemplo, en enero pasado, Doudna y Melanie Ott del Gladstone Institute of Virology en San Francisco, California, demostraron una prueba rápida CRISPR-Cas13 basada en un hisopo nasal para la detección sin amplificación del SARS-CoV-2 usando un teléfono móvil. cámara18 _
Los procedimientos de amplificación de ARN pueden aumentar la sensibilidad para rastrear secuencias virales, y Sabeti y sus colegas han desarrollado un sistema de microfluidos que detecta múltiples patógenos en paralelo utilizando material genético amplificado de solo unos pocos microlitros de muestra 19 . “En este momento, tenemos un ensayo para hacer 21 virus simultáneamente por menos de US$10 por muestra”, dice ella. Sabeti y sus colegas han desarrollado herramientas para la detección basada en CRISPR de más de 169 virus humanos a la vez, agrega.
Otras enzimas Cas podrían desarrollar la caja de herramientas de diagnóstico, señala Doudna, incluidas las proteínas Cas12, que exhiben propiedades similares a Cas13 pero se dirigen al ADN en lugar del ARN. Colectivamente, estos podrían detectar una gama más amplia de patógenos, o incluso permitir un diagnóstico eficiente de otras enfermedades no infecciosas. “Eso podría ser muy útil si pudiera hacerlo relativamente rápido, especialmente cuando los diferentes subtipos de cáncer se definen por tipos particulares de mutaciones”, dice Doudna.
Referencias
1.
Nurk, S. et al. Preimpresión en bioRxiv https://doi.org/10.1101/2021.05.26.445798 (2021).
2.
Miga, KH et al. Naturaleza 585 , 79–84 (2020).
3.
Logsdon, GA et al. Naturaleza 593 , 101–107 (2021).
4.
Gershman, A. et al. Preimpresión en bioRxiv https://doi.org/10.1101/2021.05.26.443420 (2021).
5.
Jumper, J. et al. Naturaleza 596 , 583–589 (2021).
6.
Tunyasuvunakool, K. et al. Naturaleza 596 , 590–596 (2021).
7.
Evans, R. et al. Preimpresión en bioRxiv https://doi.org/10.1101/2021.10.04.463034 (2021).
8.
Nakane, T. et al. Naturaleza 587 , 152–156 (2020).
9.
Yip, KM, Fischer, N., Paknia, E., Chari, A. y Stark, H. Nature 587 , 157–161 (2020).
10
Bernien, H. et al. Naturaleza 551 , 579–584 (2017).
11
Barredo, D., Lienhard, V., de Léséleuc, S., Lahaye, T. & Browaeys. A. Nature 561 , 79–82 (2018).
12
Jang, H. et al. Naturaleza Biomédica. Ing . https://doi.org/10.1038/s41551-021-00788-9 (2021).
13
Anzalone, AV et al. Preimpresión en bioRxiv https://doi.org/10.1101/2021.11.01.466790 (2021).
14
Levy, JM et al. Naturaleza Biomédica. Ing. 4 , 97–110 (2020).
15.
Cheng, Q. et al. Naturaleza Nanotecnología. 15 , 313–320 (2020).
16.
Liu, Y. et al. Celda 183 , 1665–1681 (2020).
17
Deng, Y. et al. Preimpresión en bioRxiv https://doi.org/10.1101/2021.06.06.447244 (2021).
18
Fozouni, P. et al. Celda 184 , 323–333 (2021).
19
Ackerman, CM et al. Naturaleza 582 , 277–282 (2020).

